
“
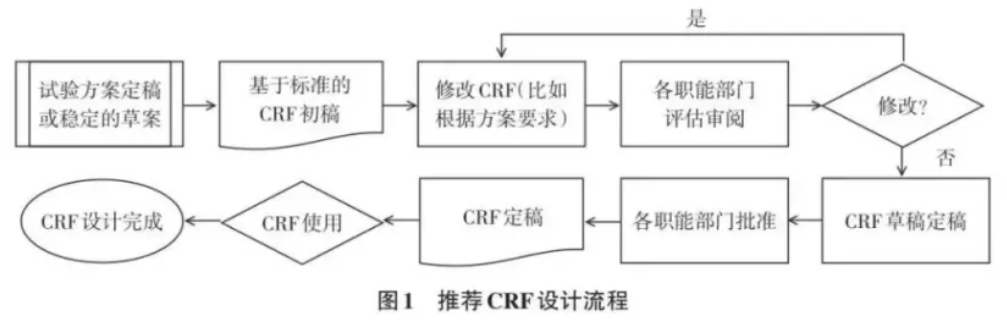
CRF设计的一般流程CRF设计一般经过初稿、修改与审查、批准与修订三个环节,设计中的每一个环节都须充分考虑其在使用中和数据处理中对后续数据管理和统计分析工作的影响,因此需要临床研究团队中的多方配合共同完成。
①先确定经审批的临床试验方案(或较为完善的方案初稿),后根据数据标准设计CRF初稿;
②CRF初稿完善:
③临床研究团队多方评阅CRF初稿并提出修改建议;
④经商讨如确定修改,则返回执行前步骤②、③,如不需要修改则定稿初CRF;
⑤经临床研究团队多方审核并批准CRF;
⑥将批准的CRF用于临床研究的数据收集;
⑦如需应研究中具体要求对CRF进行更改,则返回至步骤②执行,如不需要更改则CRF开发完成。
CRF在设计时应注意以下几个方面:
1. 符合法规及相关标准:
2. 与临床试验方案相一致:
3. 易于理解,便于实施:
4. 简明扼要,避免重复一般来说,CRF只收集与分析有关的数据,多余或重复的变量需要研究者和监查员花费时间和精力去填写和审核,这可能会减少对关键变量的注意力,影响整体数据的质量。
参考文献:
[1]卜擎燕,熊宁宁,邹建东,等.从临床研究数据管理角度设计病例报告表[J].中国新药杂志,2007,16(5):
[2]姜慧勇, 娄冬华. 临床试验既往病史、不良事件和合并用药数据清理的方法[J]. 南京医科大学学报(自然科学版), 2018, 38(10):139-142.

发表评论 取消回复